Season’s Greetings
Season’s Greetings from Sheard Scientific to all our friends and clients. Wishing a very Merry Christmas and Happy New Year to all.

Direct Reduced Iron – one of the most unpleasant cargoes out there
It really is. But why?
This article will, I hope, give you some idea why. What it won’t do is catalogue the different types of DRI you might encounter and what the requirements are to handle or carry them safely. That might be a future article, but this one is meant to explain the science underlying DRI and what gives rise to its hazards.
Iron is a very familiar substance – we make all sorts of things out of it or things based on it. Steel structures are found in buildings, cars are made from steel, we have iron railings and gates, and there are many many more examples. There’s a property in common with many of these man made objects, and that is that they rust if you don’t prevent it. Rusting is a very familiar example of an oxidation reaction. Iron plus oxygen becomes iron oxide, one type of which is the familiar red/brown rust we see on iron railings and the steelwork of older cars.

There is lots of iron around in the earth’s crust – that is because the familiar isotopes of iron are, in nuclear physics terms, the most stable substances known. Nuclear stability arises from the lack of tendency to undergo nuclear fission or nuclear fusion. Those two processes release energy from the nucleus when that nucleus splits (fission) or merges with another (fusion). Splitting a nucleus of iron will always cost energy, similarly trying to fuse iron with other nuclei will also absorb energy. Thus iron nuclei are highly stable, and form a substantial proportion of the end-products of the explosions known as supernovae and in other stellar processes. As these are where the elements making up the earth (including those in you and me!) are formed, the presence of large amounts of iron (and also nickel) in the earth’s crust and core is unsurprising. But I digress.
Iron is a moderately reactive metal and is always found in nature combined with other substances as ores. Iron ore might contain iron oxides, iron sulphides, or other forms.
Production of metallic iron from naturally occurring iron ores was first carried out by mankind over 3000 years ago. It was such an important development that the term “iron age” describes the period when iron became available for use. Early furnaces could not reach the melting point of iron and produced a form of the metal known as a “bloom”. Later furnaces used forced air draughts to reach higher temperatures and are known as blast furnaces. These produce molten iron, known as “pig” iron which can then be used in steelmaking.
Ironically (geddit?) DRI has more in common with obsolete bloom iron than more modern blast furnace forms. Consider an ore which is mostly iron oxide. Chemically, this is an example of an ionic material. The iron atoms are present as positively charged iron ions, and the oxygen atoms are present as negatively charged ions. In a piece of solid iron ore, between each pair of adjacent iron atoms is found an oxygen atom, and vice versa.
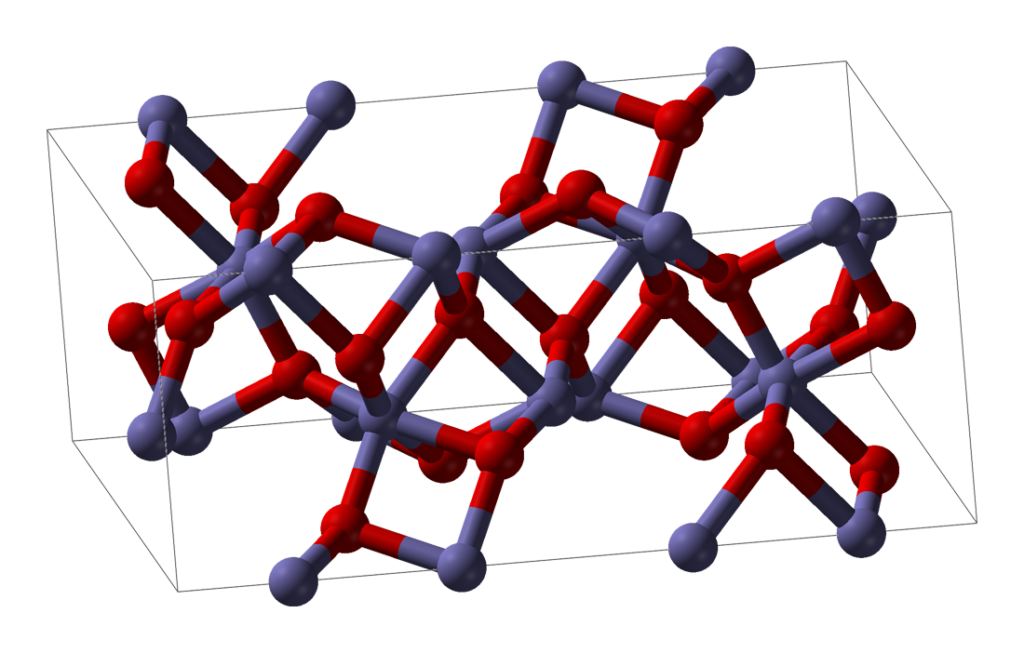
Direct reduction involves the removal of the oxygens without melting the feedstock. Typically the feedstock is pelletised iron ore, each pellet being roughly 1cm in diameter. When the oxygen is removed from the pellet without melting the pellet, the shape and size is retained but the locations which used to have oxygen atoms in them now have gaps. These tiny gaps are all interconnected and the resulting pellet of DRI is extremely porous. It has a huge surface area yet weighs less than the pellet it was made from. Once reduced, it looks very much like a malteser (without the chocolate coating).
I’m going to refer to DRI made by the MIDREX process, which is made in a shaft furnace. The reducing agent (i.e. the material used to remove the oxygen) is hydrogen and carbon monoxide made from natural gas. Natural gas is also used to heat the furnace, but because the ore is not melted, less heat is required than other furnace types, meaning that the DRI process is relatively energy-efficient. The need for natural gas to heat the furnace and provide the reducing gas means that DRI plants are usually found in locations where gas is plentiful and relatively inexpensive.
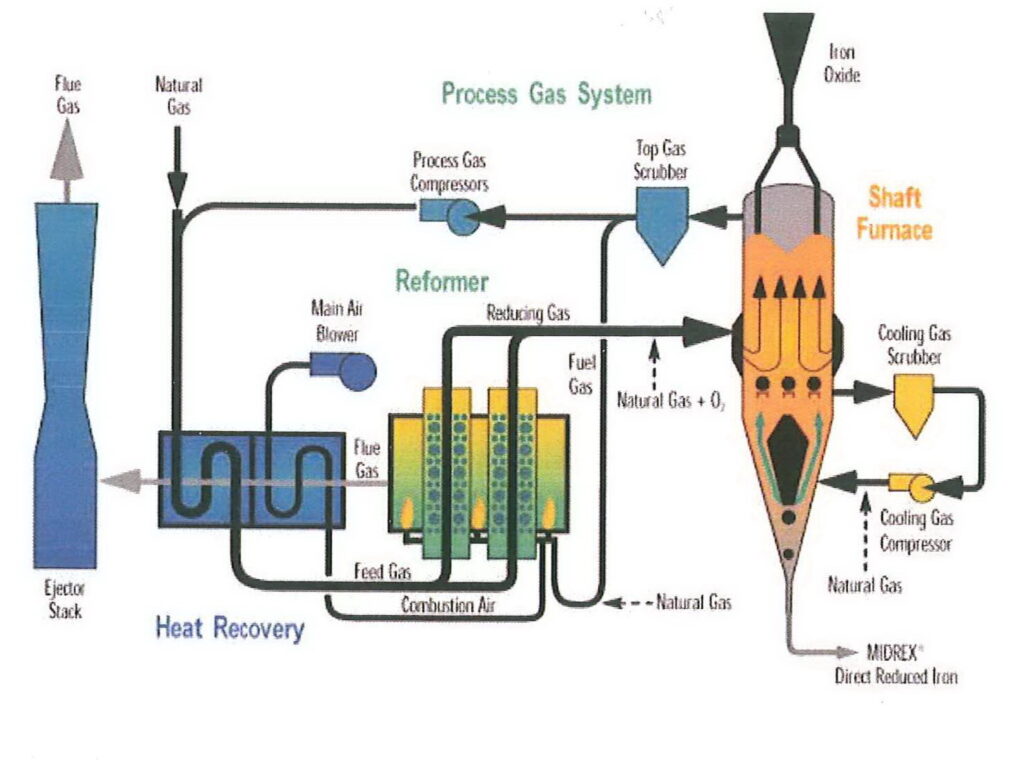
A parameter often quoted in the context of DRI is % metallisation. This is the percentage of the iron in the pellet which is present as free metallic iron rather than oxides. With modern DRI facilities, metallisation will be 95% or so. This iron can be used directly in steelmaking.
Because DRI production tends to take place where the natural gas is, it is often then carried by sea to a steel mill. That is unfortunately where some of the problems arise.
All of the problems associated with DRI come about because of the physical form that iron is in. A lump of iron can rust at its surface. That rusting usually involves water, and rusting by sea water is more rapid than with fresh water. Rusting to a cargo of DRI, especially if wetted by sea water ingress, can damage the cargo by allowing parts of it to revert to iron oxide. Loss of cargo value is undoubtedly something a shipowner will want to avoid, but there are much more immediate problems a vessel is likely to face if DRI becomes damaged on board.

Rusting is an oxidation reaction, and like all oxidative reactions, involves the release of heat. Quite a lot of heat in chemical terms, but we don’t notice a rusty car or iron railing getting hot as it rusts. Why not?
The answer is that the rusting only happens at the exposed surface of the railings or car structure, and does so slowly enough that any heat generated is immediately lost to the atmosphere. There’s never a temperature rise.
In contrast, a pellet of DRI has a huge surface area – some estimates suggest that a pellet of DRI has a surface area 10000 greater than a lump of solid iron. That means that when the DRI is freshly produced it has 10000 times the capability of reacting with oxygen and rusting. That’s one reason that DRI behaves differently to a lump or a gate made of iron.
The other hugely significant property of DRI is that, like most bulk cargoes, it is a poor conductor of heat. That might be a little surprising to hear as there is nothing there other than iron, and metals conduct heat, don’t they?
A block of iron does indeed conduct heat. A bulk made up of spheres made out of iron does not conduct very well because the contact area between adjacent spheres is small and thermal contact is poor. A bulk made up of DRI, in which much of the surface area is space rather than metal conducts heat very badly indeed.
That means that where (for example), sea water enters a bulk stow of DRI, the first thing which happens is that the wetted iron rusts. Fresh water will cause rusting but sea water makes it happen much more rapidly. The heat released by the rusting reaction, which for most of the forms of iron we encounter everyday is lost immediately, does not get conducted away and makes the temperature rise locally where the DRI was wetted.
As the temperature of the iron at those points within the stow rises, it may reach a high enough temperature that the iron can react directly with the oxygen in the hold (if there is any!). This isn’t rusting any more but is direct oxidation. The higher the temperature gets, the more rapidly the reaction goes.
If there is a ready supply of fresh air/oxygen, a DRI cargo can heat to several hundred degrees Celsius and will keep going. Whilst the rate of heat conduction within the bulk is low, it isn’t nil, and gradually the volume of DRI involved with what is now referred to as a “metal fire” increases. It may pass from hold to hold through a bulkhead. It may overheat fuel in adjacent tanks.

As I said, this article is not going to go into detail about what the different types of DRI are and how they should be carried. I will say, however, that one of the main tools in the arsenal of a vessel carrying DRI is the ability to exclude oxygen from the stow. This may be enough to prevent a fire in the first place, or it may help in keeping a situation under control so the vessel can safely reach a port where the problem can be dealt with. Nitrogen gas is used to inert holds carrying some types of DRI precisely for this purpose. Unfortunately, the more commonly found carbon dioxide inert gas is not suitable.
When DRI gets to high temperatures, it becomes a very difficult commodity to deal with. Hot DRI really loves oxygen, and will do almost anything to get it. Nitrogen is effective in stabilising where the hatches are closed, but oxygen will inevitably get into the stow during discharge. Temperatures can get very high indeed and there can be a risk to the vessel itself.
The only recourse available to assist during the discharge of a problem DRI cargo is to use water. Even that is fraught with difficulty. There are multiple problems. Firstly, problematic DRI can be at several hundreds of degrees or more. Initial amounts of water will simply be boiled off as steam. You have to be able to put water in there quicker than the DRI can boil it off.

Next problem is that water is H2O, and that O is of course oxygen. Hot DRI can satisfy its need for oxygen directly from the water you are pouring on it. The DRI takes the O, creating more heat and leaving the H2. This is hydrogen gas, very volatile and highly flammable. Be ready for whooshes of flame from the hydrogen, assuming you successfully avoid an explosion.
And then there is the fact that you are putting water, possibly even sea water, onto DRI. Any thought you might have had of salvaging any of the cargo is probably history by now, but there is a very real problem of creating further heating in stockpiles (hopefully ashore) from wetting by extinguishing water.
If all of that wasn’t enough, the final insult is that that the resulting oxidised DRI will weigh about 30% more than the DRI cargo you started with. Anecdotally, problems involving the collapse of quay structures have been caused by underestimating the weight of the reoxidised cargo.
Phew. If you are going to carry DRI, it really is very important that you don’t have a problem. But DRI can cause difficulties even when there isn’t an emergency/fire situation. What might they be?
Inevitably during loading and discharge, dust is created. That dust is metallic iron. The IMSBC Code warns about the need to remove accumulations of dust as quickly as possible. I have seen first-hand what happens if you don’t or can’t. Accumulations of DRI dust embed themselves into paint and then rust. This is not only unsightly but it can cause significant operational problems. The Code also warns about the potential for damage to radio and other aerials/antennae.
All in all, carriage of DRI is not something to be embarked upon without a great deal of preparation and care. At Sheard Scientfic, we have first-hand experience of dealing with carriage and consequences of carriage of DRI and can assist our clients with this highly problematic material.
A lasting feat of engineering

For as long as I can remember, the Settle-Carlisle railway line and the stations it served have been threatened by closure. This includes, famously, the amazing Ribblehead Viaduct. Begun in 1869, it was a necessary part of the Midland Railway’s project to have their own route to Scotland. A fabulous sight, whether from Ribblehead Station, from underneath the viaduct, whether it is being traversed by modern rolling stock or British Railways 46100 Royal Scot with a rake of historic Pullman coaches.




Biotechnology and GMOs – Part 2 – what is PCR?
In recent weeks, a number of P&I Clubs have circulated an information document kindly produced by Istanbul Correspondents Vitsan. This document warns that Turkish Courts have banned the import of distillers dried grains & solubles (DDGS) cargoes containing a particular genetically modified maize variety (MON810, more about that later).

The document warns that paperwork needs to be in order regarding shipments of DDGS being imported to Turkey. It seems likely that some form of testing on arrival may take place, and that is going to use a test known as PCR. But what is it?
PCR is the Polymerase Chain Reaction. There you go – shortest post ever!
Ah, you want to know how it works and what it does? OK, step this way. All through the weekly COVID Government updates, we heard the experts and Ministers use this acronym repeatedly. Before the advent of cheap lateral flow tests, it was the only way to establish if someone had that infection. It remains the way of investigating which variant of the virus an individual has, demonstrating when new variants appear, and tracking the spread of those variants. Whenever you see DNA testing referred to on CSI/forensics TV shows, PCR is the technique they are applying.
As a tool, it has revolutionised molecular biology, and it is difficult to imagine modern genetics being developed without it. That is remarkable bearing in mind it was only developed in the 1980s.
There’s no substitute to understanding the basics of how it works. Fundamentally, it selectively reproduces and amplifies a specific segment of DNA, if it is present.
I discussed in a previous post what DNA is and how it codes for information. DNA exists as a double-helix, with the gaps between the backbones occupied by base pairs AT, TA, GC and CG. This is referred to as double-stranded DNA. Each sugar backbone molecule (dexoyribose sugar) is joined to the next sugar in the same way – carbon 5 on one molecule is joined to carbon 3 on the next. The two helices run in the opposite directions and are joined together by the base pairs.
When DNA is heated up, the two strands separate. This is sometimes called “melting”, and it happens at a temperature of around 94-98oC. The two strands, now separated from each other, both contain the full genetic information the DNA coded for. That is because each A on the top strand was originally paired with a T on the bottom, each G with a C and so on.
This means that if there was a mechanism of adding new bases and deoxyribose sugars to a single strand of DNA, the full double stranded molecule could be recreated. This is what happens in cellular division in real life – the double-stranded DNA is separated into two single strands (not by heat in a cell, though!) and the cell then creates new matching strands leaving it with two identical DNA molecules. This replication process is where errors can arise resulting in genetic mutations, but this article isn’t the place to go into that.
So, inevitably, cells already have the equipment to synthesise a replacement strand to convert single-stranded DNA to double-stranded. This equipment is an enzyme known as a DNA polymerase. It needs a supply of free bases and dexoyribose sugar backbones to work.
It also needs something else. DNA polymerase requires a segment of double-stranded DNA in order to start. Once it has started, it will continue literally up the existing strand of DNA, manufacturing a complementary strand to produce double stranded DNA. Think of it as being like a zipper – you have to start a zipper by aligning the starting pieces. DNA polymerase works along like a zipper but it manufactures one side of the zip as it goes.
In a PCR reaction, short segments of single stranded DNA sequences known as primers are put into the reaction vessel. These are designed to be a perfect match for part of the DNA being tested for. Two primers are used – one matches a sequence on one strand of DNA, the other matches a sequence on the other.
In order for the primers to settle and bind on the sequences they match, the temperature has to be lower than the 94-98oC I mentioned above to separate double-stranded DNA. Typically this happens around 50-65oC. The primer sequences then act as the starter for the DNA polymerase to start and zip from.
Most DNA polymerase enzymes are not sufficiently thermally stable to survive being heated to 94-98oC in the first step. Such enzymes would need to be added, and then the “zipping”/elongation stage is usually run around 72-75oC. However, a crucial discovery for the development of PCR was the existence of a DNA polymerase from the organism Thermus aquaticus, which can survive at the 94-98oC required to denature DNA. This enzyme is known as Taq polymerase after the organism it was extracted from.
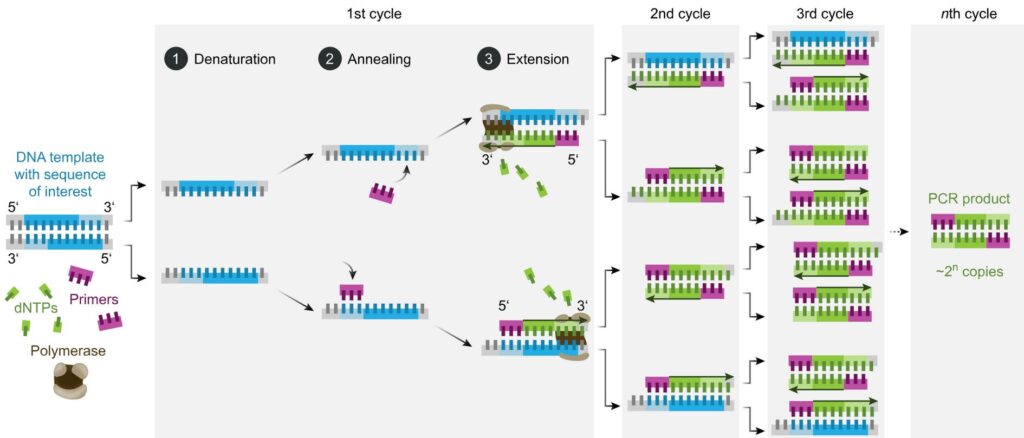
PCR is a reaction run in cycles. A reaction vessel is used into which are placed the DNA to be tested, primer sequences, base pairs on deoxyribose sugar backbones, and Taq polymerase. The reaction proceeds as follows (leaving out initial and final temperature changes which are significant but outside the scope of this article).
- Denaturation. 94-98oC for 20-30 seconds. Long enough to separate the double-stranded DNA to single stranded but not long enough to destroy the Taq enzyme.
- Annealing. 50-65oC for 20-40 seconds. In this time the primers bind to DNA sequences on the target DNA, if those sequences are present. Choice of primers and temperature are crucial – see below.
- Elongation. 72oC. Required time varies. In this period the Taq polymerase synthesises a new strand of DNA starting at a primer and running onwards until it reaches the end of the DNA strand or runs out of time.
- Repeat
The reaction is controlled by the temperature – thus the reaction tubes are placed in a machine which cycles the temperature through the regime listed above. Each full cycle takes a couple of minutes. In an hour or so, it is straightforward for 30 cycles to be run without any more reactants needing to be added to the vessel or any other intervention.
As mentioned above, PCR is used to amplify in a selective manner a particular fragment of DNA. Assume that we are testing for the presence of MON810 maize DNA, and that the sample being tested does indeed contain that DNA – but only one molecule of it. (Potentially bad news if you are importing that maize to Turkey). The denaturation step will create two single-stranded segments of DNA from the extracted maize sample. These will not be the same but one will be effectively the reverse/mirror of the other. One primer must be designed to be a match/complement to a DNA sequence on one strand, and the other primer to a different area on the second strand. They bracket the DNA sequence to be amplified.
On the first cycle, one primer will bind to each strand of the sample DNA, and those will be elongated in step 3. Note that the DNA sections extracted from the maize sample might be very long indeed. The elongated copies made in this first step 3 will start at a primer (because that is how Taq polymerase works) and will extend for some distance until the point where the polymerase ran out of time in step 3.
On the second cycle things become interesting. Denaturation will produce four single-strands of DNA. Two of these will be the original ones from the sample (of whatever length they were) but the other two will start with one of the primers and will then be of random length. Call the two primers P1 and P2.
The strand synthesised in the first reaction starting with P1 doesn’t contain a sequence P1 can bind to – so only P2 can bind. Similarly, the strand which started with P2 in the first cycle will then bind with P1. Elongation in the second cycle will produce double sided DNA from all four single strands. Two of these (the ones produced from the synthesised segments in cycle 1) will have a new segment synthesised which starts at P1 and stops at P2. That is the desired amplification product.
At the end of the second cycle, we are left with four double stranded segments of DNA. When denatured at the start of the third cycle, six of these (i.e. the two original DNA strands, the two synthesised in cycle 1, and two of the four synthesised in cycle 2) are what is known as variable length strands. The other two however are exactly the same length as each other, and that length is the number of DNA base pairs between the sequences in the original DNA which bind to P1 and P2.
This might not sound so great, as we have three times as many DNA fragments of variable length than the specific sequence we were intending to multiply.
However, at the end of this third cycle, we have 16 strands of DNA. Eight of these are variable length fragments (two new variable length fragments were synthesised in cycle 3 from the original sample DNA). However, the other eight are exactly the same length – each starts at the location of P1 and runs to the location of P2.
At the end of the fourth cycle we have 32 strands of DNA. Ten are variable length, but all of the rest are exactly the same – starting at P1 and ending at P2.
However, now run the reaction cycle a further 29 times. You end up with a couple of hundred variable length fragments and over 1,000,000,000 (one US billion) identical copies of a fragment exactly the length bounded by P1 and P2. The enormous quantities of exact length fragments completely swamp the variable lengths fragments.
The resulting contents of the reaction tube are then visualised in some way so that the results can be understood. They might be separated on a gel – this is how the charts are produced showing the characteristic lines we see in DNA results. Each line is an amplified fragment – a fragment bounded by two specific primers. These amplified fragments, even from a tiny original amount of DNA, are present in such enormous numbers they are readily detected and visualised.
To return to our example, if the sample of maize DNA didn’t contain a fragment from MON810, that fragment couldn’t be amplified, and that “line” would be absent on the chart.
Everything then depends on the selection of suitable primers (and to some extent, the fine-tuning of the temperatures used in the PCR cycles). The primers need to be long enough that there is no chance of those sequences being found elsewhere in the maize genome, and they need to be short enough to bind with the target DNA during step 2 of each cycle – if they don’t then there is nothing for Taq polymerase to zip from.
As an example, the EU reference test for MON810 uses the following primers
- P1 – TCGAAGGACGAAGGACTCTAACGT
- P2 – GCCACCTTCCTTTTCCACTATCTT (reverse strand)
This test is known as an “event specific” test because primer P1 binds to a sequence on the maize parent plant whereas P2 binds to a sequence on the inserted gene – i.e. the fragment to be amplified contains a section of DNA from the maize and a section from the insert, it flanks the genetic insertion. Thus this test only detects MON810, it doesn’t (for instance) detect other GMOs which contain the same inserted DNA.
More on the use of PCR to detect GMOs in relation to shipping in a later article.
For now, I will offer some comments on the generalities of using PCR and the MON810 variation.
The enormous specific amplification provided by even 30 cycles of PCR means that very tiny amounts of DNA can be detected. Provided the primers are appropriately selected, the test is wholly specific – it only detects the sequences it was designed to detect. There are off-the-shelf primer kits available for commercially produced GMO plants. Thus an event specific test can be anticipated to exist for any GMO likely to be found in a real world shipment.
As long ago as 1999, methods for detection of GMOs were being studied in the context of commercial commodities. This is because of the fear which has existed for some time of “Frankenstein foods” and other issues relating to escape of modified material into the natural environment. One particular study looked for a specific GMO soya type (commonly known as Roundup-Ready soya) not in samples of soya but in wheat bread. Soya was only in the bread as a minor component of the baking improver/powder. Despite the steps making up the dough production and baking, GMO soya was readily detected in loaves produced using that baking aid – even though the soya made up only 0.4% of the dough mix. In that study, commercial baking aids were also tested, and in 2 out of 15 off-the-shelf products tested the GMO soya could be detected in the baked bread. Back in 1999!
And so, by a roundabout route we return to maize and MON810. MON810 is quite an early GMO product of the Monsanto company. It was made by inserting DNA from a bacterium Bacillus thuringiensis. In the bacterium this produces a toxin which affects certain butterflies and moths, some of which as larvae damage maize plants. The idea of the modification was that the maize plants would produce the toxin, and that would serve as a control over the larvae, thus reducing the need to apply pesticides. MON810 has been commercially very successful and has been very widely planted. The modification was produced by firing projectiles containing the bacterial DNA at plant cells.
MON810 has been the subject of some controversy. It has been suggested that the use of this variety can be responsible for the death of bee colonies. As far as I am aware, this allegation, or indeed any other specific allegations of harmful effects from MON810 has not been proven.
As stated above, the EU test for MON810 is event specific and will detect very small amounts of the GMO. Even a fragment from a MON810 plant will, if it ends up in a sample sent for testing, lead to a positive result. I note that the Vitsan circular relates to imports of distillers dried grains & solubles (DDGS) – not the maize itself. DDGS is produced from harvested maize by a series of industrial steps, but by analogy to the baking powder/bread study referenced above, these processes are not likely to prevent detection of modified genetic material if MON810 was present in the original unprocessed maize.
I note that Vitsan have specifically clarified that a different product, NK603xMON810 maize is allowed to be imported, provided it is referenced as MON-00603-6xMON-00810-6. The “x” in this designation identifies that this is a so-called “stacked trait”. This means that the maize variety MON-00603-6xMON-00810-6 carries the properties of MON810 and the properties of MON603. The GMO event 603 is a Roundup-Ready variety of maize. Thus the stacked trait is resistant to the herbicide Roundup (glyphosate) and also produces the bacterial toxin to kill the larvae mentioned above.
Often, but not always, stacked traits are produced by conventional genetic crossing techniques. If this is done, and plants which express both traits are selected for, then BOTH events will be present in the marketed product. Thus a test for event MON810 will be positive, as will a test for event 603. It is not clear to the writer what undesirable properties could exist in MON810 which are not also present in MON-00603-6xMON-00810-6. I do not have sufficient data yet to say whether MON-00603-6xMON-00810-6 will test positive in an event specific test for MON810.
It appears likely that there may be difficulties with testing surrounding verification for this regulation – i.e. cargoes of DDGS may be rejected when they should not be. The presence of genetically modified material is not something which can be detected by the naked eye nor even field testing. It requires sampling and laboratory analysis, and there is always scope for a misleading result.