In recent weeks, a number of P&I Clubs have circulated an information document kindly produced by Istanbul Correspondents Vitsan. This document warns that Turkish Courts have banned the import of distillers dried grains & solubles (DDGS) cargoes containing a particular genetically modified maize variety (MON810, more about that later).

The document warns that paperwork needs to be in order regarding shipments of DDGS being imported to Turkey. It seems likely that some form of testing on arrival may take place, and that is going to use a test known as PCR. But what is it?
PCR is the Polymerase Chain Reaction. There you go – shortest post ever!
Ah, you want to know how it works and what it does? OK, step this way. All through the weekly COVID Government updates, we heard the experts and Ministers use this acronym repeatedly. Before the advent of cheap lateral flow tests, it was the only way to establish if someone had that infection. It remains the way of investigating which variant of the virus an individual has, demonstrating when new variants appear, and tracking the spread of those variants. Whenever you see DNA testing referred to on CSI/forensics TV shows, PCR is the technique they are applying.
As a tool, it has revolutionised molecular biology, and it is difficult to imagine modern genetics being developed without it. That is remarkable bearing in mind it was only developed in the 1980s.
There’s no substitute to understanding the basics of how it works. Fundamentally, it selectively reproduces and amplifies a specific segment of DNA, if it is present.
I discussed in a previous post what DNA is and how it codes for information. DNA exists as a double-helix, with the gaps between the backbones occupied by base pairs AT, TA, GC and CG. This is referred to as double-stranded DNA. Each sugar backbone molecule (dexoyribose sugar) is joined to the next sugar in the same way – carbon 5 on one molecule is joined to carbon 3 on the next. The two helices run in the opposite directions and are joined together by the base pairs.
When DNA is heated up, the two strands separate. This is sometimes called “melting”, and it happens at a temperature of around 94-98oC. The two strands, now separated from each other, both contain the full genetic information the DNA coded for. That is because each A on the top strand was originally paired with a T on the bottom, each G with a C and so on.
This means that if there was a mechanism of adding new bases and deoxyribose sugars to a single strand of DNA, the full double stranded molecule could be recreated. This is what happens in cellular division in real life – the double-stranded DNA is separated into two single strands (not by heat in a cell, though!) and the cell then creates new matching strands leaving it with two identical DNA molecules. This replication process is where errors can arise resulting in genetic mutations, but this article isn’t the place to go into that.
So, inevitably, cells already have the equipment to synthesise a replacement strand to convert single-stranded DNA to double-stranded. This equipment is an enzyme known as a DNA polymerase. It needs a supply of free bases and dexoyribose sugar backbones to work.
It also needs something else. DNA polymerase requires a segment of double-stranded DNA in order to start. Once it has started, it will continue literally up the existing strand of DNA, manufacturing a complementary strand to produce double stranded DNA. Think of it as being like a zipper – you have to start a zipper by aligning the starting pieces. DNA polymerase works along like a zipper but it manufactures one side of the zip as it goes.
In a PCR reaction, short segments of single stranded DNA sequences known as primers are put into the reaction vessel. These are designed to be a perfect match for part of the DNA being tested for. Two primers are used – one matches a sequence on one strand of DNA, the other matches a sequence on the other.
In order for the primers to settle and bind on the sequences they match, the temperature has to be lower than the 94-98oC I mentioned above to separate double-stranded DNA. Typically this happens around 50-65oC. The primer sequences then act as the starter for the DNA polymerase to start and zip from.
Most DNA polymerase enzymes are not sufficiently thermally stable to survive being heated to 94-98oC in the first step. Such enzymes would need to be added, and then the “zipping”/elongation stage is usually run around 72-75oC. However, a crucial discovery for the development of PCR was the existence of a DNA polymerase from the organism Thermus aquaticus, which can survive at the 94-98oC required to denature DNA. This enzyme is known as Taq polymerase after the organism it was extracted from.
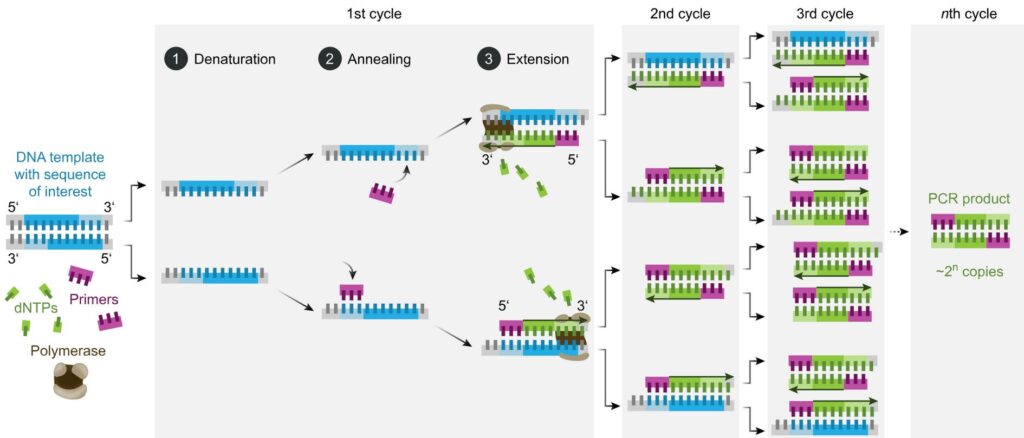
PCR is a reaction run in cycles. A reaction vessel is used into which are placed the DNA to be tested, primer sequences, base pairs on deoxyribose sugar backbones, and Taq polymerase. The reaction proceeds as follows (leaving out initial and final temperature changes which are significant but outside the scope of this article).
- Denaturation. 94-98oC for 20-30 seconds. Long enough to separate the double-stranded DNA to single stranded but not long enough to destroy the Taq enzyme.
- Annealing. 50-65oC for 20-40 seconds. In this time the primers bind to DNA sequences on the target DNA, if those sequences are present. Choice of primers and temperature are crucial – see below.
- Elongation. 72oC. Required time varies. In this period the Taq polymerase synthesises a new strand of DNA starting at a primer and running onwards until it reaches the end of the DNA strand or runs out of time.
- Repeat
The reaction is controlled by the temperature – thus the reaction tubes are placed in a machine which cycles the temperature through the regime listed above. Each full cycle takes a couple of minutes. In an hour or so, it is straightforward for 30 cycles to be run without any more reactants needing to be added to the vessel or any other intervention.
As mentioned above, PCR is used to amplify in a selective manner a particular fragment of DNA. Assume that we are testing for the presence of MON810 maize DNA, and that the sample being tested does indeed contain that DNA – but only one molecule of it. (Potentially bad news if you are importing that maize to Turkey). The denaturation step will create two single-stranded segments of DNA from the extracted maize sample. These will not be the same but one will be effectively the reverse/mirror of the other. One primer must be designed to be a match/complement to a DNA sequence on one strand, and the other primer to a different area on the second strand. They bracket the DNA sequence to be amplified.
On the first cycle, one primer will bind to each strand of the sample DNA, and those will be elongated in step 3. Note that the DNA sections extracted from the maize sample might be very long indeed. The elongated copies made in this first step 3 will start at a primer (because that is how Taq polymerase works) and will extend for some distance until the point where the polymerase ran out of time in step 3.
On the second cycle things become interesting. Denaturation will produce four single-strands of DNA. Two of these will be the original ones from the sample (of whatever length they were) but the other two will start with one of the primers and will then be of random length. Call the two primers P1 and P2.
The strand synthesised in the first reaction starting with P1 doesn’t contain a sequence P1 can bind to – so only P2 can bind. Similarly, the strand which started with P2 in the first cycle will then bind with P1. Elongation in the second cycle will produce double sided DNA from all four single strands. Two of these (the ones produced from the synthesised segments in cycle 1) will have a new segment synthesised which starts at P1 and stops at P2. That is the desired amplification product.
At the end of the second cycle, we are left with four double stranded segments of DNA. When denatured at the start of the third cycle, six of these (i.e. the two original DNA strands, the two synthesised in cycle 1, and two of the four synthesised in cycle 2) are what is known as variable length strands. The other two however are exactly the same length as each other, and that length is the number of DNA base pairs between the sequences in the original DNA which bind to P1 and P2.
This might not sound so great, as we have three times as many DNA fragments of variable length than the specific sequence we were intending to multiply.
However, at the end of this third cycle, we have 16 strands of DNA. Eight of these are variable length fragments (two new variable length fragments were synthesised in cycle 3 from the original sample DNA). However, the other eight are exactly the same length – each starts at the location of P1 and runs to the location of P2.
At the end of the fourth cycle we have 32 strands of DNA. Ten are variable length, but all of the rest are exactly the same – starting at P1 and ending at P2.
However, now run the reaction cycle a further 29 times. You end up with a couple of hundred variable length fragments and over 1,000,000,000 (one US billion) identical copies of a fragment exactly the length bounded by P1 and P2. The enormous quantities of exact length fragments completely swamp the variable lengths fragments.
The resulting contents of the reaction tube are then visualised in some way so that the results can be understood. They might be separated on a gel – this is how the charts are produced showing the characteristic lines we see in DNA results. Each line is an amplified fragment – a fragment bounded by two specific primers. These amplified fragments, even from a tiny original amount of DNA, are present in such enormous numbers they are readily detected and visualised.
To return to our example, if the sample of maize DNA didn’t contain a fragment from MON810, that fragment couldn’t be amplified, and that “line” would be absent on the chart.
Everything then depends on the selection of suitable primers (and to some extent, the fine-tuning of the temperatures used in the PCR cycles). The primers need to be long enough that there is no chance of those sequences being found elsewhere in the maize genome, and they need to be short enough to bind with the target DNA during step 2 of each cycle – if they don’t then there is nothing for Taq polymerase to zip from.
As an example, the EU reference test for MON810 uses the following primers
- P1 – TCGAAGGACGAAGGACTCTAACGT
- P2 – GCCACCTTCCTTTTCCACTATCTT (reverse strand)
This test is known as an “event specific” test because primer P1 binds to a sequence on the maize parent plant whereas P2 binds to a sequence on the inserted gene – i.e. the fragment to be amplified contains a section of DNA from the maize and a section from the insert, it flanks the genetic insertion. Thus this test only detects MON810, it doesn’t (for instance) detect other GMOs which contain the same inserted DNA.
More on the use of PCR to detect GMOs in relation to shipping in a later article.
For now, I will offer some comments on the generalities of using PCR and the MON810 variation.
The enormous specific amplification provided by even 30 cycles of PCR means that very tiny amounts of DNA can be detected. Provided the primers are appropriately selected, the test is wholly specific – it only detects the sequences it was designed to detect. There are off-the-shelf primer kits available for commercially produced GMO plants. Thus an event specific test can be anticipated to exist for any GMO likely to be found in a real world shipment.
As long ago as 1999, methods for detection of GMOs were being studied in the context of commercial commodities. This is because of the fear which has existed for some time of “Frankenstein foods” and other issues relating to escape of modified material into the natural environment. One particular study looked for a specific GMO soya type (commonly known as Roundup-Ready soya) not in samples of soya but in wheat bread. Soya was only in the bread as a minor component of the baking improver/powder. Despite the steps making up the dough production and baking, GMO soya was readily detected in loaves produced using that baking aid – even though the soya made up only 0.4% of the dough mix. In that study, commercial baking aids were also tested, and in 2 out of 15 off-the-shelf products tested the GMO soya could be detected in the baked bread. Back in 1999!
And so, by a roundabout route we return to maize and MON810. MON810 is quite an early GMO product of the Monsanto company. It was made by inserting DNA from a bacterium Bacillus thuringiensis. In the bacterium this produces a toxin which affects certain butterflies and moths, some of which as larvae damage maize plants. The idea of the modification was that the maize plants would produce the toxin, and that would serve as a control over the larvae, thus reducing the need to apply pesticides. MON810 has been commercially very successful and has been very widely planted. The modification was produced by firing projectiles containing the bacterial DNA at plant cells.
MON810 has been the subject of some controversy. It has been suggested that the use of this variety can be responsible for the death of bee colonies. As far as I am aware, this allegation, or indeed any other specific allegations of harmful effects from MON810 has not been proven.
As stated above, the EU test for MON810 is event specific and will detect very small amounts of the GMO. Even a fragment from a MON810 plant will, if it ends up in a sample sent for testing, lead to a positive result. I note that the Vitsan circular relates to imports of distillers dried grains & solubles (DDGS) – not the maize itself. DDGS is produced from harvested maize by a series of industrial steps, but by analogy to the baking powder/bread study referenced above, these processes are not likely to prevent detection of modified genetic material if MON810 was present in the original unprocessed maize.
I note that Vitsan have specifically clarified that a different product, NK603xMON810 maize is allowed to be imported, provided it is referenced as MON-00603-6xMON-00810-6. The “x” in this designation identifies that this is a so-called “stacked trait”. This means that the maize variety MON-00603-6xMON-00810-6 carries the properties of MON810 and the properties of MON603. The GMO event 603 is a Roundup-Ready variety of maize. Thus the stacked trait is resistant to the herbicide Roundup (glyphosate) and also produces the bacterial toxin to kill the larvae mentioned above.
Often, but not always, stacked traits are produced by conventional genetic crossing techniques. If this is done, and plants which express both traits are selected for, then BOTH events will be present in the marketed product. Thus a test for event MON810 will be positive, as will a test for event 603. It is not clear to the writer what undesirable properties could exist in MON810 which are not also present in MON-00603-6xMON-00810-6. I do not have sufficient data yet to say whether MON-00603-6xMON-00810-6 will test positive in an event specific test for MON810.
It appears likely that there may be difficulties with testing surrounding verification for this regulation – i.e. cargoes of DDGS may be rejected when they should not be. The presence of genetically modified material is not something which can be detected by the naked eye nor even field testing. It requires sampling and laboratory analysis, and there is always scope for a misleading result.